La gestione dell’acromegalia richiede il coinvolgimento di un team di specialisti: endocrinologi, neurochirurghi, radioterapisti, dietisti, psicologi e altri esperti che possano seguire ogni aspetto della patologia
L’acromegalia è una patologia rara che si manifesta in età adulta a causa di una produzione eccessiva di ormone della crescita (GH). Si stima che colpisca tra i 60 e i 120 individui ogni milione di persone. L’anomalia ormonale si verifica dopo la pubertà, quando la crescita in altezza è ormai conclusa poiché le cartilagini di coniugazione delle ossa si sono saldate. Il GH in eccesso non potendo più agire sull’accrescimento in altezza, stimola invece l’ingrossamento di ossa, cartilagini, tessuti molli e organi.

Acromegalia in una donna, foto di repertorio
Differenze con il gigantismo
Nel caso in cui l’eccesso di ormone della crescita si manifesti prima della pubertà, la condizione risultante è il gigantismo, caratterizzato da un’anomala crescita staturale. L’acromegalia, a differenza, comporta un aumento volumetrico delle estremità corporee e di altri tessuti, poiché lo sviluppo verticale è ormai impossibile.
La principale causa dell’acromegalia è un adenoma ipofisario, un tumore benigno che si sviluppa nell’ipofisi, una ghiandola situata alla base del cervello nella sella turcica, una cavità ossea dell’osso sfenoide. Questo tumore, nella maggior parte dei casi, è in grado di secernere ormoni in maniera incontrollata. Più raramente, la malattia può dipendere da tumori localizzati in altri organi come i polmoni, il pancreas o le ghiandole surrenali.
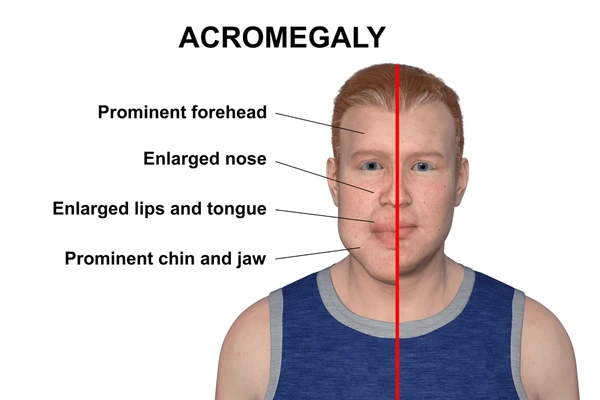
Il termine “acromegalia” deriva dal greco akros (estremità) e megalos (grande), e fa riferimento all’ingrandimento di mani, piedi e altre parti distali del corpo. Questo sintomo caratteristico è spesso il primo indizio che porta alla diagnosi.
L’acromegalia si sviluppa lentamente e i segni possono restare inosservati per anni. In genere, tra l’inizio della malattia e la diagnosi trascorrono da 5 a 10 anni. Tra i sintomi più comuni vi sono:
- Aumento della taglia di mani e piedi (con necessità di cambiare indumenti e monili come guanti, scarpe, anelli);
- Modificazioni del volto: crescita sproporzionata della mandibola (prognatismo), ingrossamento del naso, degli zigomi e delle labbra;
- Aumento della lingua (macroglossia);
- Ispessimento della pelle e dei tessuti molli;
- Sudorazione eccessiva (iperidrosi), spesso accompagnata da odore acre;
- Voce più profonda, anche nelle donne;
- Cefalea, stanchezza cronica e dolori articolari;
- Sindrome del tunnel carpale;
- Disturbi visivi, ipertensione, apnee notturne e diabete.
L’ormone della crescita influisce anche sul metabolismo, inducendo insulino-resistenza, iperglicemia e, in circa un terzo dei pazienti, diabete mellito. Questo rende fondamentale un controllo metabolico continuo.
Acromegalia sintomi e diagnosi. L’acromegalia rappresenta una sfida clinica per la complessità delle sue complicanze, che spaziano da alterazioni metaboliche e cardiovascolari a problemi oncologici e neurologici. L’eccesso di GH e IGF-I, insieme alla pressione meccanica esercitata dagli adenomi ipofisari, determina un impatto significativo sulla salute generale. Una gestione efficace richiede un monitoraggio continuo e un trattamento personalizzato, supportati da evidenze scientifiche che sottolineano l’importanza di interventi precoci per mitigare i rischi e migliorare la prognosi
Complicanze
Le complicanze dell’acromegalia si dividono in due categorie principali:
- Complicanze ormonali: coinvolgono l’intero organismo, con ispessimento di organi interni (cuore, fegato, reni, tiroide, pancreas), aumentato rischio di tumori (noduli tiroidei, polipi del colon), ipertensione, problemi cardiaci e disfunzioni ormonali multiple.
- Complicanze anatomiche: causate dalla crescita dell’adenoma, che può comprimere strutture cerebrali vicine come il chiasma ottico (riduzione del campo visivo) o i nervi cranici (causando paralisi o dolori facciali). In alcuni casi, l’adenoma o il suo trattamento compromettono la produzione di altri ormoni ipofisari vitali, come il cortisolo o gli ormoni sessuali, con conseguenze potenzialmente gravi (amenorrea, osteoporosi, infertilità, ipotiroidismo centrale e diabete insipido).
Questo squilibrio ormonale causa alterazioni metaboliche e proliferative che si manifestano con complicanze sistemiche e locali, spesso legate alla presenza di un adenoma ipofisario. La gestione di tali complicanze richiede un approccio integrato e una diagnosi tempestiva per ridurre i rischi e migliorare la qualità di vita dei pazienti. Di seguito, un’analisi sintetica delle principali conseguenze della malattia, supportata da evidenze cliniche.
L’ipersecrezione di GH e IGF-I provoca un’ampia gamma di alterazioni sistemiche, influenzando diversi apparati. Tra le complicanze metaboliche, l’iperglicemia e l’intolleranza glucidica sono frequenti, con circa il 30% dei pazienti che sviluppa diabete mellito di tipo 2. Questo è dovuto alla capacità del GH di antagonizzare l’azione dell’insulina, portando a un’alterata regolazione del glucosio. Studi clinici hanno dimostrato che l’eccesso di GH aumenta la gluconeogenesi epatica e riduce la sensibilità insulinica periferica, contribuendo a un profilo metabolico simile a quello della sindrome metabolica.
Il sistema cardiovascolare è particolarmente vulnerabile. L’acromegalia è associata a un rischio doppio di mortalità per cause cardiache, con un’incidenza significativa di ipertrofia miocardica, cardiomiopatia dilatativa, aritmie e aterosclerosi coronarica. L’ispessimento del miocardio, rilevato in oltre il 50% dei pazienti con malattia attiva, è spesso accompagnato da disfunzione diastolica e, in fasi avanzate, da insufficienza cardiaca. Ricerche pubblicate su riviste come The Lancet Endocrinology evidenziano che l’ipertensione arteriosa, presente in circa il 40% dei casi, amplifica ulteriormente il danno cardiovascolare.
Un’altra complicanza rilevante è la sindrome delle apnee ostruttive del sonno (OSAS), che colpisce fino al 70% dei pazienti. L’ispessimento dei tessuti molli del faringe, causato dalla proliferazione indotta dal GH, ostruisce le vie aeree superiori, portando a episodi di apnea notturna che compromettono la qualità del sonno e aumentano il rischio cardiovascolare. Inoltre, l’effetto proliferativo del GH è stato associato a un incremento del rischio oncologico. Studi epidemiologici, come quelli pubblicati su Endocrine Reviews, indicano una maggiore prevalenza di polipi intestinali, gozzo nodulare e, in misura minore, tumori maligni al colon e alla tiroide, sebbene la causalità diretta rimanga controversa.
Complicanze Locali da Adenoma Ipofisario
L’acromegalia è spesso causata da un adenoma ipofisario, che può generare complicanze locali in base alle sue dimensioni e alla sua espansione. I macroadenomi (>1 cm) possono comprimere strutture circostanti, con effetti significativi. Quando l’adenoma si estende verso il chiasma ottico, può causare disturbi visivi, come l’emianopsia bitemporale, dovuta alla compressione delle fibre nervose ottiche. Studi di neuroimaging, come la risonanza magnetica, mostrano che circa il 20% dei pazienti con macroadenomi sviluppa deficit visivi significativi.
L’invasione laterale verso i seni cavernosi può coinvolgere i nervi cranici III, IV, V e VI, causando sintomi come diplopia, ptosi palpebrale o dolore facciale. Inoltre, l’espansione dell’adenoma può compromettere la funzione ipofisaria, portando a ipopituitarismo. La carenza di ormoni come ACTH, TSH, FSH e LH può provocare ipocortisolismo, ipotiroidismo centrale, infertilità o amenorrea, con conseguenti effetti sistemici come stanchezza cronica, osteoporosi e riduzione della libido. In rari casi, l’interferenza con la neuroipofisi può indurre diabete insipido, caratterizzato da poliuria e sete intensa, con un rischio significativo di disidratazione se non trattato.
Approccio Clinico e Importanza della Diagnosi Precoce
Le complicanze dell’acromegalia, sia sistemiche che locali, sottolineano la necessità di un approccio multidisciplinare che coinvolga endocrinologi, neurochirurghi, radiologi e altri specialisti. La diagnosi precoce, attraverso dosaggi di GH e IGF-I e imaging ipofisario, è cruciale per prevenire l’evoluzione delle complicanze. Studi clinici, come quelli pubblicati su Journal of Clinical Endocrinology & Metabolism, dimostrano che un trattamento tempestivo, che include chirurgia transfenoidale, terapie farmacologiche (es. analoghi della somatostatina) o radioterapia, può ridurre significativamente il rischio di morbilità e mortalità. Inoltre, il controllo delle comorbilità, come il diabete e l’ipertensione, è essenziale per migliorare gli esiti a lungo termine.
Diagnosi dell’Acromegalia
Il percorso diagnostico inizia con il dosaggio nel sangue dell’IGF-1 (somatomedina C), il mediatore dell’azione del GH, che presenta valori stabili e affidabili nel tempo. In caso di risultati elevati, si effettua il test da carico orale di glucosio: nei soggetti sani, il GH si abbassa dopo l’assunzione di glucosio, mentre nei pazienti con acromegalia resta alto. In presenza di valori patologici, si procede con una risonanza magnetica con contrasto per visualizzare l’ipofisi e l’adenoma.
Il trattamento dell’acromegalia ha l’obiettivo di riportare i livelli di GH e IGF-1 alla normalità, alleviare i sintomi e prevenire le complicanze.
- Chirurgia: la prima opzione è la rimozione dell’adenoma attraverso un intervento transfenoidale (passando dal naso). Questo approccio è minimamente invasivo e risolutivo nella maggioranza dei microadenomi e in circa metà dei macroadenomi.
- Farmaci: se l’intervento non è possibile o risolutivo, si utilizzano farmaci come:
- Analoghi della somatostatina (octreotide, lanreotide), che riducono la secrezione di GH;
- Antagonisti del recettore del GH (pegvisomant), che bloccano l’azione dell’ormone;
- Agonisti della dopamina (cabergolina), meno efficaci ma utili in alcuni casi.
- Radioterapia: riservata a casi resistenti alla chirurgia e alla terapia farmacologica. La tecnica stereotassica consente una precisione elevata, minimizzando i danni ai tessuti sani.
I pazienti con acromegalia devono adottare abitudini alimentari equilibrate e uno stile di vita attivo, soprattutto per contrastare il rischio cardiovascolare, l’ipertensione e il diabete.
Dal punto di vista medico, per arrestare la progressione dell’acromegalia, la strategia più efficace è nella rimozione del tumore ipofisario, generalmente benigno, che ne è la causa. L’intervento chirurgico, spesso eseguito con tecnica transfenoidale attraverso il naso, permette di accedere e rimuovere il tumore. Ci sono dei casi in cui un residuo tumorale non asportabile chirurgicamente può persistere, causando una continua sovrapproduzione di ormone della crescita: in queste situazioni si ricorre a trattamenti farmacologici o radioterapia per controllare l’eccesso ormonale e prevenire la crescita del tumore residuo. La scelta del trattamento e il suo tempismo vengono definiti individualmente attraverso valutazioni interdisciplinari.
L’acromegalia è una malattia subdola, che può portare a conseguenze gravi se non diagnosticata e trattata in tempo. Riconoscere i segni iniziali, come l’aumento di mani e piedi o cambiamenti del volto, è fondamentale per un intervento tempestivo. La disponibilità di opzioni terapeutiche efficaci e la presenza di centri multidisciplinari specializzati permettono oggi una gestione ottimale della malattia, migliorando l’aspettativa e la qualità della vita dei pazienti.